DAL “W ALE NOTIZIE” N°38, DEL 15 DICEMBRE 2019)
Rubrica Scientifica a cura del Prof. Cosmoferruccio De Stefano
La diagnosi differenziale deve peraltro tenere conto che il linfedema è presente anche in quadri sindromici complessi come la sindrome di Turner 45,X), la sindrome di Noonan, la sindrome di Mucke e di Prader-Willi.Tra le forme più note di linfedema primitivo, citiamo la sindrome di Meige, denominata anche “linfedema-distichiasi”.
Il linfedema è tipicamente localizzato agli arti inferiori, l’esordio è tardivo, generalmente durante la pubertà e si associa a distichiasi (cioè la crescita delle ciglia con direzione anomala, verso la cornea invece che all’esterno). La distichiasi, già presente alla nascita, può esser causa di irritazioni corneali, congiuntiviti ricorrenti e fotofobia e si accompagna a metaplasia squamosa delle ghiandole di Meibonio.
Come la malattia di Milroy, la malattia di Meige si trasmette con carattere autosomico dominante ad alta penetranza ed espressività variabile ed il gene principalmente coinvolto è rappresentato dal “forhead transcription factors” (FOXC2,) che normalmente regola lo sviluppo vascolare dei vasi linfatici.
La terza forma di linfedema primitivo sin ad oggi conosciuta, è rappresentata dalla sindrome “Ipotricosi-linfedema-telengectasia”. Essa si differenzia dalle altre forme, per la perdita dei capelli, l’insorgenza di linfedema durante l’infanzia e le telengectasie distribuite prevalentemente al palmo delle mani. La trasmissione della malattia può avvenire con modalità autosomica dominante o recessiva. Tutti i pazienti che presentano la malattia di Milroy, vengono peraltro sottoposti a indagini relativamente invasive quali la linfoangiografia e la linfoscintigrafia per la loro accuratezza diagnostica. Sottoponendo i pazienti a linfoscintigrafia, si è osservato che il drenaggio linfatico e la funzione dei vasi linfatici degli arti era completamente normale, tuttavia, a livello dello stravaso di liquidi, la linfoscintigrafia dimostrava un’alterazione dell’assorbimento linfatico. Contrariamente a quanto si pensava, la malattia non è causata da un’aplasia dei vasi linfatici, bensì da una loro insufficienza funzionale. Tale osservazione è stata confermata dopo aver sottoposto i pazienti ad accurate indagini, come la biopsia cutanea e l’ecografia-ecocolordoppler dei vasi, da cui emerge che i vasi linfatici non sono funzionanti e che esiste un’alta prevalenza di reflusso venoso superficiale.
Il linfedema può riscontrarsi all’interno di sindromi collegate al non corretto numero di cromosomi. Esempi di patologie dovute ad anomalie dei cromosomi sessuali sono: la sindrome di Klinefelter e la sindrome di Turner.
La prima è dovuta alla presenza di un cromosoma X in più (XXY al posto di XY), 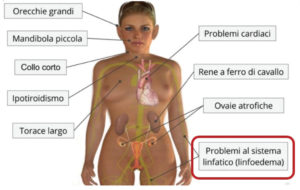 e colpisce i maschi con una frequenza di un caso su 700 nuovi nati; la sindrome di Turner invece è causata dalla mancanza di un cromosoma X (X anziché XX), ed interessa le femmine con una frequenza di 1 caso su 2500 nuove nate.
e colpisce i maschi con una frequenza di un caso su 700 nuovi nati; la sindrome di Turner invece è causata dalla mancanza di un cromosoma X (X anziché XX), ed interessa le femmine con una frequenza di 1 caso su 2500 nuove nate.
Le monosomie complete sono incompatibili con la vita postnatale, l’eccezione è rappresentata dalla monosomia del cromosoma X, associata alla sindrome di Turner (45,X). Le caratteristiche fenotipiche associate alla sindrome di Turner sono elencate in figura.
In epoca neonatale, nella sindrome di Turner, spesso è visibile il Linfedema delle estremità degli arti superiori e inferiori (puffiness).
Un ultimo capitolo legato alle cause che sono alla base del linfedema è rappresentato dalle sindromi dismorfiche ad eredità multifattoriale quali:
− Woltman (linfedema con enteropatia proteino- disperdente).
− Linfangectasie polmonari congenite.
− Hennekam.
− Idrope fetale non immune.
− Linfedema-ipoparatiroidismo.
− Linfedema-microcefalia-corioretinoptia.
− Sindrome di Noonan.
La sindrome di Noonan (SN) è caratterizzata da cardiopatia congenita, bassa statura, pterigio del collo, anomalie del torace, criptorchidismo, difetti di coagulazione, anomalie linfatiche e dismorfie facciali tipiche.
A livello del sistema linfatico la SN è caratterizzata da Ipoplasia di vasi linfatici può condurre a linfedema del dorso delle mani e dei piedi o, più raramente, a Linfangectasie intestinali, polmonari o testicolari, versamenti chiusi nello spazio pleurico e nel peritoneo. Già in epoca prenatale possono essere osservati igroma cistico o idrope fetale.
Nella sindrome di Noonan, la diagnosi clinica può essere confermata nel 75% dei pazienti mediante una correlazione genotipo fenotipo. Nel 50% dei casi il gene coinvolto è PTPN11. Le caratteristiche fenotipiche sono rappresentate da bassa statura, stenosi polmonare valvolare e dismorfismi facciali. Nel 10% dei casi il gene coinvolto è SOS1. Le caratteristiche fenotipiche sono rappresentate da accrescimento staturo ponderale normale, assenza di difetto cognitivo, presenza di anomalie cutanee, predisposizione a tumori, cherubinismo, sinovite villodulare. Nel 5% dei casi il gene coinvolto è SHOC2. Le caratteristiche fenotipiche sono rappresentate da anomalie del capello (loose anagen hair) e cutanee associate a deficit di GH. Nel 3-7% dei casi il gene coinvolto è RAF1. Le caratteristiche fenotipiche sono rappresentate da cardiomiopatia ipertrofica. In una irrisoria casistica la sindrome di Noonan è causata da mutazioni nei geni KRAS, BRAF, MAP2K1 e CBL.
Concludendo, oltre al riconoscimento della causa genetica del linfedema è importante, dal punto di vista clinico, l’utilizzo della ecografia e della linfoscintirafia, la prima è una indagine di prima scelta per la diagnosi ed il trattamento del linfedema, la seconda svolge un ruolo importante nella diagnosi e prevenzione del linfedema.
Domenico Dell’Edera
Rosalba Ardea Dell’Edera
Maria Teresa Dell’Edera